También llamado Mucopolisacaridosis VI (MPS VI), es una enfermedad metabólica hereditaria, clasificada como de depósito lisosomal, causada por la ausencia o el mal funcionamiento de una enzima llamada arilsulfatasa B (ASB), la cual es necesaria para la degradación de unas moléculas en las células llamadas glicosoaminoglicanos (GAGs) o mucopolisacáridos, como eran llamadas anteriormente. Estos GAGs son largas cadenas de azúcares, presentes en cada una de las células del cuerpo, y hacen parte del colágeno presente en muchos órganos como los cartílagos, huesos, tendones, córneas, piel y el tejido conjuntivo.
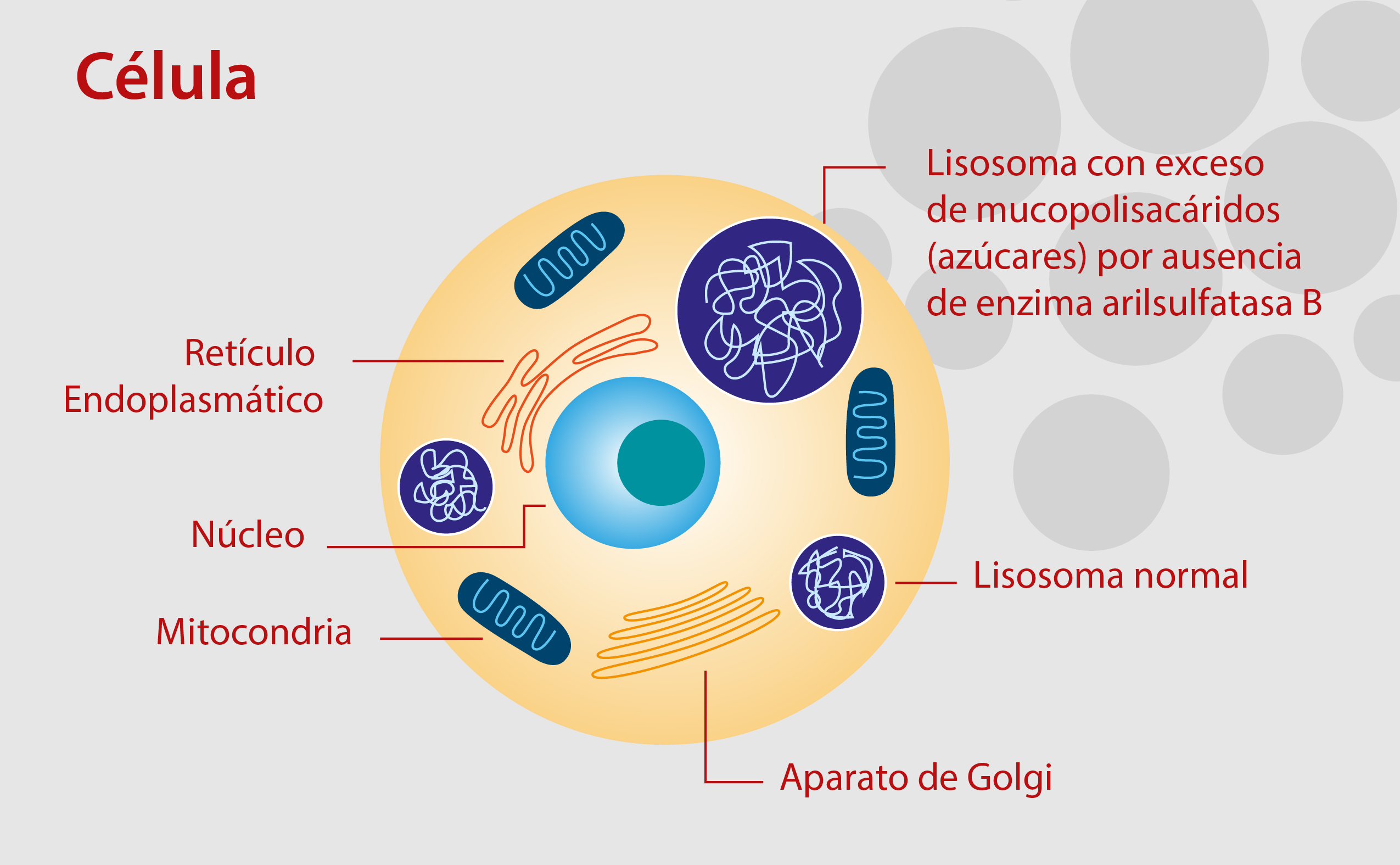

Debido a este déficit de la ASB, el exceso de glicosaminoglicanos se acumulan dentro de los lisosomas en muchos órganos del cuerpo; entre ellos la piel, el corazón, las vías respiratorias y el esqueleto; ocasionando síntomas generalizados.
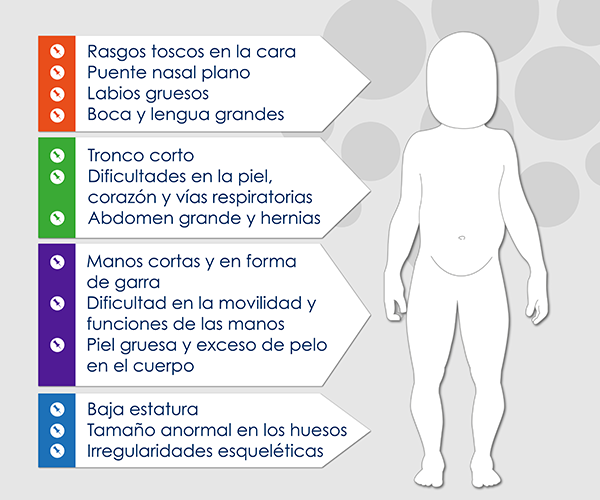
Los pacientes evidencian rasgos toscos en la cara, baja estatura, un puente nasal plano, unos labios gruesos, boca y lengua grandes. Así mismo, un tronco corto, un tamaño anormal de los huesos y otras irregularidades esqueléticas. La piel es gruesa y con crecimiento excesivo de pelo en el cuerpo. El abdomen puede ser grande debido a un crecimiento anormal del hígado y el bazo. Es común encontrar hernias. Las manos son cortas y en forma de garra. Debido a la rigidez de las coyunturas y el síndrome de túnel del carpo se dificultan la movilidad y las funciones de la mano.
Fuente informativa: scp.com.co/precop-old/precop_files/modulo_4_vin_3/mucopolisacaridosis.pdf
http://www.mpsesp.org/portal1/default.asp#
Versión Escrita: Fundaper